
We focus on two broad aspects of understanding the limits on bacterial growth: translation rate and accuracy, and the fitness consequences of new mutations. An important requirement for cellular growth is the ability to rapidly and reliably produce large amounts of proteins. In bacteria, fast growth is thought to impose strong selection for rapid translation, influencing key aspects of the process such as codon use, tRNA content, and rRNA copy number. On the other hand, translation also needs to be accurate, because high levels of mistranslation reduce the amount of functional protein available in the cell; and mistranslated proteins can be toxic. Hence, variation in key genomic features relevant for translation may directly or indirectly determine bacterial translation and growth.
Translation rate and accuracy
From early sequencing data it was clear that different genes and genomes preferentially use different sets of codons. These patterns are thought to reflect selection acting to maximize translation speed, and/or minimize translation errors. However, the fitness consequences of codon use, and mechanisms underlying these effects, remained poorly understood for decades. This is important to understand, because large fitness consequences of codon changes (i.e. synonymous mutations) would imply that a fundamental assumption of molecular evolution – that synonymous mutations are neutral – is incorrect.
We showed that synonymous codon changes in an essential protein-coding gene can have major impacts on bacterial growth (Agashe et al. 2013; Agashe et al. 2016). However, contrary to expectations from the translational selection hypothesis, we found that the fitness effects were not explained by codon use per se. Instead, the local sequence context is critical and determines the unique (and thus far unpredictable) effect of each synonymous change. As others have pointed out, apart from directly changing translation speed or accuracy, codon changes may also alter mRNA structure, alter co-translational protein folding dynamics, or generate sequence motifs that hinder translation (e.g. internal Shine Dalgarno (SD)-like motifs that can stall ribosomes). We found that none of these mechanisms explained the magnitude of fitness effects of synonymous mutations, suggesting multiple and complex underlying mechanisms.

In fact, across ~300 sequenced bacterial genomes, we did not find evidence supporting the hypothesis that purifying selection to avoid ribosomal stalling depletes SD-like motifs in bacterial mRNAs (Diwan and Agashe 2016). On the contrary, we found that such motifs are enriched in the 3’ ends of mRNAs, potentially serving as ribosomal binding sites for downstream genes. Thus, although specific synonymous mutations can have major impacts on bacterial fitness, our work suggests that growth is not predictably and directly dependent on codon use or transcriptional sequence motifs. This conclusion is also supported by our recent bioinformatics analysis demonstrating that tRNA genes and codon use show distinct, amino acid-specific responses to faster growth, and may co-evolve only under a narrow range of growth rates (Mahajan and Agashe 2018). Together, our work indicates that altered codon use may generally have very weak effects on translation rate or accuracy, and may significantly limit bacterial growth only under specific conditions.
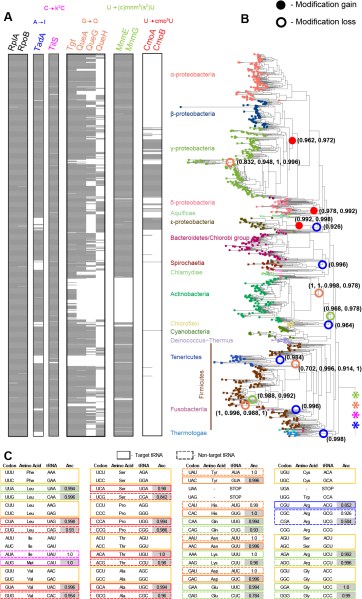
Compared to codon use, tRNA pools may have a more direct influence on translation speed or accuracy, given their central role in the process of translation. Therefore, we investigated factors affecting the tRNA repertoire of bacteria. Typically, bacteria only encode 26–45 types of tRNAs, which is not sufficient to decode all 61 sense codons. This shortfall is met by tRNA modifying enzymes that allow specific tRNAs to more efficiently and accurately decode multiple (rather than single) codons. Thus, in the presence of tRNA modifications, a minimal set of tRNAs could effectively decode all codons, suggesting that tRNA modifications may have played a major role in the evolution of the genetic code and tRNA pools. However, the evolution and impact of bacterial tRNA modifications has not been systematically analyzed.
We traced the evolutionary history and role of tRNA modifying enzymes across a phylogeny of 1093 bacteria, showing that most modifications were ancient, but were lost repeatedly in different lineages (Diwan and Agashe 2018). These losses were often associated with altered tRNA diversity driven by shifts in GC content and codon use, or with genome reduction. We propose that the altered tRNA diversity weakened selection on tRNA modifications, allowing their loss through drift. In turn, the loss of modifications should generate strong selection to retain the expanded tRNA set. Parth is now experimentally testing some of the predictions arising from this work.
The broader perspective on the evolution and impact of codon use and tRNA pools is shaped by the assumption of strong selection to maximize translation accuracy. However, mistranslation rates are very high in most organisms, and the evolutionary impacts of mistranslation are relatively poorly understood. Focusing on initiator tRNA mutants in E. coli (which show increased mistranslation rates), Laasya and Parth have tested the short- and long-term evolutionary effects of mistranslation, and the underlying molecular mechanisms.
In our work, genomic GC content emerged as the ultimate driver of multiple aspects of translation, contradicting the prevalent idea that key translational components – tRNA genes, codon use, or rRNA genes – ultimately constrain bacterial growth. Although bacteria show enormous intra- and inter-genomic variation in GC content, this variability has not been analysed comprehensively (Agashe and Shankar 2014). Hence, Saurabh used phylogenetic approaches to analyze the evolutionary processes that drive major shifts in bacterial genomic GC content.
